






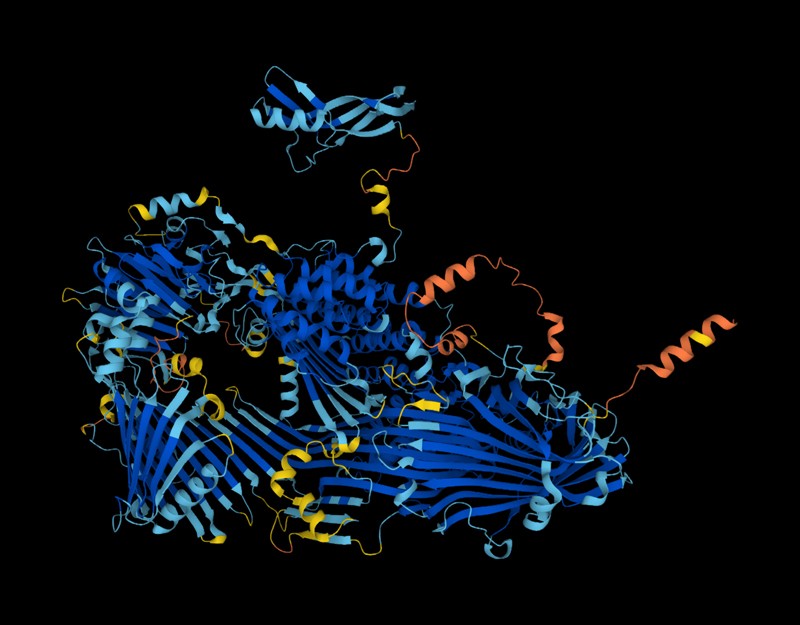
Die Struktur des Vitellogenin-Proteins – ein Vorläufer von Eigelb – wie vom AlphaFold-Tool vorhergesagt.Bildnachweis: DeepMind
Ab heute ist die Bestimmung der 3D-Form fast aller wissenschaftlich bekannten Proteine so einfach wie das Eintippen einer Google-Suche.
Die Forscher nutzten AlphaFold – das revolutionäre Netzwerk für künstliche Intelligenz (KI) – um die Strukturen von rund 200 Millionen Proteinen aus 1 Million Arten vorherzusagen, die fast alle bekannten Proteine auf dem Planeten abdecken.
Der Daten-Dump wird in einer Datenbank frei verfügbar sein, die von DeepMind, dem in London ansässigen KI-Unternehmen von Google, das AlphaFold entwickelt hat, und dem European Molecular Biology Laboratory’s European Bioinformatics Institute (EMBL-EBI), einer zwischenstaatlichen Organisation in der Nähe von Cambridge, Großbritannien, eingerichtet wurde.
„Im Wesentlichen kann man sich vorstellen, dass es das gesamte Proteinuniversum abdeckt“, sagte Demis Hassabis, CEO von DeepMind, auf einer Pressekonferenz. „Wir stehen am Beginn einer neuen Ära der digitalen Biologie.“
Die 3D-Form oder -Struktur eines Proteins bestimmt seine Funktion in Zellen. Die meisten Medikamente werden anhand von Strukturinformationen entwickelt, und genaue Karten sind oft der erste Schritt zur Entdeckung der Funktionsweise von Proteinen.
DeepMind hat das AlphaFold-Netzwerk mit einer KI-Technik namens Deep Learning entwickelt, und die AlphaFold-Datenbank wurde vor einem Jahr mit 350.000 Strukturvorhersagen gestartet, die fast jedes Protein abdecken, das Menschen, Mäusen und 19 anderen umfassend untersuchten Organismen bekannt ist. Der Katalog ist seitdem auf ca. 1 Million Einträge angewachsen.
„Wir bereiten uns auf die Veröffentlichung dieses großartigen Vermögenswerts vor“, sagt Christine Orengo, eine Computerbiologin am University College London, die die AlphaFold-Datenbank zur Identifizierung neuer Proteinfamilien verwendet hat. „Alle Daten für unsere Prognose zu haben, ist einfach fantastisch.“
Hochwertige Strukturen
Die Veröffentlichung von AlphaFold im letzten Jahr sorgte für Furore in der Life-Science-Community, die Schwierigkeiten hatte, das Tool zu nutzen. Das Netzwerk erstellt hochgenaue Vorhersagen der 3D-Form oder -Struktur von Proteinen. Es liefert auch Informationen über die Genauigkeit seiner Vorhersagen, sodass die Forscher wissen, worauf sie sich verlassen können. Traditionell verwendeten Wissenschaftler zeitaufwändige und teure experimentelle Methoden wie Röntgenkristallographie und Kryo-Elektronenmikroskopie, um Proteinstrukturen aufzuklären.
Laut EMBL-EBI gelten etwa 35 % der mehr als 214 Millionen Vorhersagen als hochgenau, das heißt, sie sind so gut wie experimentell bestimmte Strukturen. Weitere 45 % wurden als zuversichtlich genug eingestuft, um sich bei vielen Anwendungen darauf verlassen zu können.
Viele AlphaFold-Strukturen sind gut genug, um experimentelle Strukturen für einige Anwendungen zu ersetzen. In anderen Fällen verwenden Forscher AlphaFold-Vorhersagen, um experimentelle Daten zu validieren und zu interpretieren. Schlechte Vorhersagen sind oft offensichtlich, und einige von ihnen werden durch eine intrinsische Störung im Protein selbst verursacht, was bedeutet, dass es keine definierte Form hat, zumindest wenn keine anderen Moleküle vorhanden sind.
Die heute veröffentlichten 200 Millionen Vorhersagen basieren auf den Sequenzen in einer anderen Datenbank namens UNIPROT. Es ist wahrscheinlich, dass Wissenschaftler bereits eine Vorstellung von der Form einiger dieser Proteine hatten, weil sie in Datenbanken mit experimentellen Strukturen enthalten sind oder anderen Proteinen in solchen Aufbewahrungsorten ähneln, sagt Eduard Porta Pardo, ein Bioinformatiker am Josep Carreras Leukemia Research Institute (IJC ) in Barcelona.
Aber solche Einträge tendieren dazu, sich auf Proteine von Menschen, Mäusen und anderen Säugetieren zu konzentrieren, sagt Porta, also ist es wahrscheinlich, dass der AlphaFold-Dump bedeutendes Wissen hinzufügen wird, weil er von viel vielfältigeren Organismen stammt. „Das wird eine großartige Ressource. Und ich werde es wahrscheinlich herunterladen, sobald es herauskommt“, sagt Porta.
Da die AlphaFold-Software seit einem Jahr verfügbar ist, waren die Forscher bereits in der Lage, die Struktur jedes gewünschten Proteins vorherzusagen. Viele sagen jedoch, dass die Verfügbarkeit von Vorhersagen in einer einzigen Datenbank den Forschern Zeit, Geld und Aufwand sparen wird. „Es ist eine weitere Eintrittsbarriere, die Sie beseitigen“, sagt Porta. „Ich habe viele AlpahFold-Modelle verwendet. Ich habe AlphaFold noch nie selbst ausgeführt.“
Jan Kosinski, Strukturmodellierer am EMBL Hamburg in Deutschland, der das AlphaFold-Netzwerk seit einem Jahr betreibt, kann die Datenbankerweiterung kaum erwarten. Sein Team verbrachte drei Wochen damit, das Proteom – die Gesamtheit aller Proteine eines Organismus – eines Krankheitserregers vorherzusagen. „Jetzt können wir einfach alle Modelle herunterladen“, sagte er während des Briefings.
Einhundert Terabyte
Fast jedes bekannte Protein in der Datenbank zu haben, wird auch neue Arten von Studien ermöglichen. Orengos Team nutzte die AlphaFold-Datenbank, um neue Arten von Proteinfamilien zu identifizieren, und sie werden dies nun in einem viel größeren Maßstab tun. Ihr Labor wird die umfangreiche Datenbank auch nutzen, um die Evolution von Proteinen mit nützlichen Eigenschaften zu verstehen, wie der Fähigkeit, Plastik zu konsumieren, oder besorgniserregenden, wie solchen, die Krebs verursachen können. Die Identifizierung entfernter Verwandter dieser Proteine in der Datenbank kann die Grundlage für ihre Eigenschaften bilden.
Martin Steinegger, ein Bioinformatiker an der Seoul National University, der an der Entwicklung einer Cloud-basierten Version von AlphaFold mitgewirkt hat, freut sich über die Erweiterung der Datenbank. Aber er sagt, dass Forscher das Netzwerk wahrscheinlich immer noch selbst verwalten müssen. Menschen verwenden AlphaFold zunehmend, um zu bestimmen, wie Proteine interagieren, und solche Vorhersagen sind nicht in der Datenbank enthalten. Mikrobielle Proteine werden auch nicht durch Sequenzierung von genetischem Material aus Boden, Meerwasser und anderen „metagenomischen“ Quellen identifiziert.
Einige anspruchsvolle Anwendungen der umfangreichen AlphaFold-Datenbank könnten auch darauf angewiesen sein, die gesamten 23 Terabyte an Inhalten herunterzuladen, was für viele Teams nicht machbar wäre, sagt Steinegger. Cloud-basierter Speicher kann auch teuer sein. Steinegger hat ein Softwaretool namens FoldSeek mitentwickelt, das strukturell ähnliche Proteine schnell finden kann und in der Lage sein soll, die AlphaFold-Daten signifikant auszudrucken.
Selbst wenn jedes bekannte Protein enthalten ist, muss die AlphaFold-Datenbank aktualisiert werden, wenn neue Organismen entdeckt werden. Die Vorhersagen von AlphaFold können sich auch verbessern, wenn neue strukturelle Informationen verfügbar werden. Hassabis sagt, DeepMind habe sich verpflichtet, die Datenbank langfristig zu unterstützen, und er könne jährliche Aktualisierungen erwarten.
Seine Hoffnung ist, dass die Verfügbarkeit der AlphaFold-Datenbank die Lebenswissenschaften nachhaltig beeinflussen wird. „Es wird ein ziemlich großes Umdenken erfordern.“

Schöpfer. Hipster-freundlicher Unternehmer. Student. Freundlicher Analyst. Professioneller Schriftsteller. Zombie-Guru. Amateur-Web-Nerd.










+ There are no comments
Add yours